Convert geneids into GRanges
genes_to_granges.RdConvert geneids into GRanges
genes_to_granges(geneids, txdb, complement = TRUE, plot = TRUE, verbose = TRUE) genefile_to_granges(file, txdb, complement = TRUE, plot = TRUE)
Arguments
| geneids | Gene identifier vector |
|---|---|
| txdb | |
| complement | TRUE (default) or FALSE: add complementary strand? |
| plot | TRUE (default) or FALSE |
| verbose | TRUE (default) or FALSE |
| file | Gene identifier file (one per row) |
Value
See also
Examples
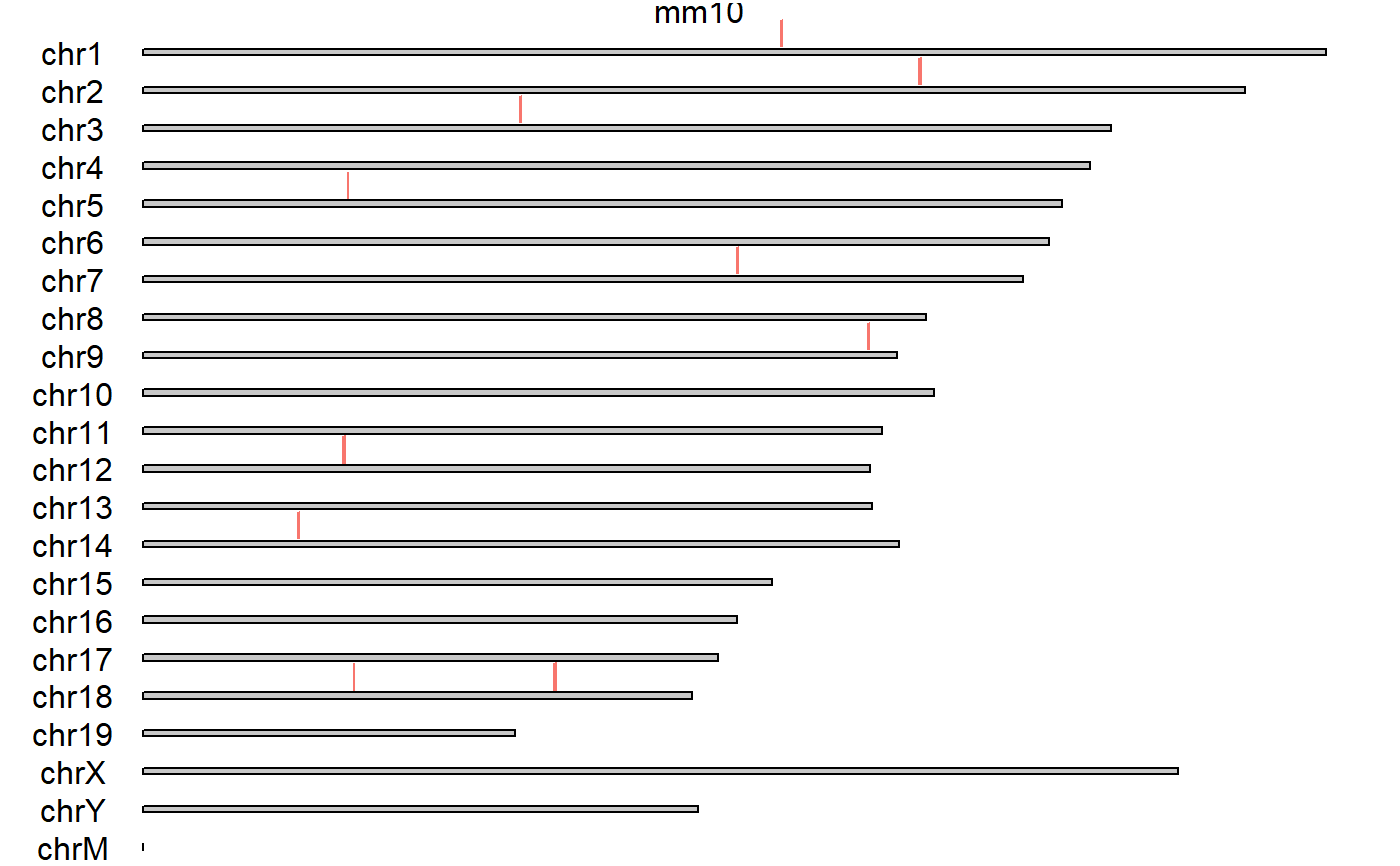
# Entrez #------- genefile <- system.file('extdata/SRF.entrez', package='multicrispr') geneids <- as.character(read.table(genefile)[[1]]) txdb <- getFromNamespace('TxDb.Mmusculus.UCSC.mm10.knownGene', 'TxDb.Mmusculus.UCSC.mm10.knownGene') (gr <- genes_to_granges(geneids, txdb))#> #> #> #> #>#>#>#> GRanges object with 20 ranges and 4 metadata columns: #> seqnames ranges strand | gene_id targetname #> <Rle> <IRanges> <Rle> | <character> <character> #> 27392_r chr1 105518422-105663677 - | 27392 27392_r #> 27392_f chr1 105518422-105663677 + | 27392 27392_f #> 100043424_r chr2 128178319-128502765 - | 100043424 100043424_r #> 100043424_f chr2 128178319-128502765 + | 100043424 100043424_f #> 622434_f chr3 62338344-62462221 + | 622434 622434_f #> ... ... ... ... . ... ... #> 71918_f chr14 25711642-25769039 + | 71918 71918_f #> 13653_f chr18 34859823-34864984 + | 13653 13653_f #> 13653_r chr18 34859823-34864984 - | 13653 13653_r #> 52662_f chr18 67933257-68268630 + | 52662 52662_f #> 52662_r chr18 67933257-68268630 - | 52662 52662_r #> targetstart targetend #> <integer> <integer> #> 27392_r 105518422 105663677 #> 27392_f 105518422 105663677 #> 100043424_r 128178319 128502765 #> 100043424_f 128178319 128502765 #> 622434_f 62338344 62462221 #> ... ... ... #> 71918_f 25711642 25769039 #> 13653_f 34859823 34864984 #> 13653_r 34859823 34864984 #> 52662_f 67933257 68268630 #> 52662_r 67933257 68268630 #> ------- #> seqinfo: 66 sequences (1 circular) from mm10 genome(gr <- genefile_to_granges(genefile, txdb))#> #> #> #> #>#>#>#> GRanges object with 20 ranges and 4 metadata columns: #> seqnames ranges strand | gene_id targetname #> <Rle> <IRanges> <Rle> | <character> <character> #> 27392_r chr1 105518422-105663677 - | 27392 27392_r #> 27392_f chr1 105518422-105663677 + | 27392 27392_f #> 100043424_r chr2 128178319-128502765 - | 100043424 100043424_r #> 100043424_f chr2 128178319-128502765 + | 100043424 100043424_f #> 622434_f chr3 62338344-62462221 + | 622434 622434_f #> ... ... ... ... . ... ... #> 71918_f chr14 25711642-25769039 + | 71918 71918_f #> 13653_f chr18 34859823-34864984 + | 13653 13653_f #> 13653_r chr18 34859823-34864984 - | 13653 13653_r #> 52662_f chr18 67933257-68268630 + | 52662 52662_f #> 52662_r chr18 67933257-68268630 - | 52662 52662_r #> targetstart targetend #> <integer> <integer> #> 27392_r 105518422 105663677 #> 27392_f 105518422 105663677 #> 100043424_r 128178319 128502765 #> 100043424_f 128178319 128502765 #> 622434_f 62338344 62462221 #> ... ... ... #> 71918_f 25711642 25769039 #> 13653_f 34859823 34864984 #> 13653_r 34859823 34864984 #> 52662_f 67933257 68268630 #> 52662_r 67933257 68268630 #> ------- #> seqinfo: 66 sequences (1 circular) from mm10 genome# Ensembl #-------- # txdb <- AnnotationHub::AnnotationHub()[["AH75036"]] # genefile <- system.file('extdata/SRF.ensembl', package='multicrispr') # geneids <- as.character(read.table(genefile)[[1]]) # (gr <- genes_to_granges(geneids, txdb)) # (gr <- genefile_to_granges(genefile, txdb))